The reference font for the body text of European proposals is Times New Roman (Windows platforms), Times/Times New Roman (Apple platforms) or Nimbus Roman No. 9 L (Linux distributions). The Roman family is from a pre-digital age and has well-recognizable features.
Is it the best font in terms of readability? On the one hand, there is a tendency to move from Times-type fonts to plainer fonts, like Calibri. On the other hand, many studies (with controversial results) account for aspects like Dyslexia, typeface anatomy, and Display vs. Print. The effect of font choice on readability and compression on big numbers seems small or insignificant. However, my point is that a proposal must be clear to a few reviewers, who might have difficulties understanding the proposal due to age, Dyslexia, and colour vision deficiency. These few people will have some feelings about how the text is formatted. For that reason and also because of my artistic education in caligraphy, I have been looking for and playing with font combinations for a long time. Here is what I have tried and liked.
1. STIX two and Source Sans form a pair of Serif and Sans fonts. STIX two resulted from a collaborative effort from the most prominent academic publishing companies. Its predecessor (STIX one) has exactly the same metrics as Times New Roman. STIX two is somewhat bigger, which is not prohibited by the EU funding agencies. The main benefit of using STIX fonts is that these are mathematical fonts and, thus, can be natively used in MS Equation Editor (instead of Cambria) and LaTeX (as XITS or STIX2).
2. An excellent substitution for Times New Roman is Zilla Slab – a unique font by the Mozilla foundation – which has the same metrics as Times New Roman, is a Sans font, yet looks like a monospace one, does have features of a Dyslexia-friendly typeface, and looks great in print and on screen. It is freely available from Google fonts. It can be used with Times New Roman (or similar) as a pair of Serif and Sans fonts.
3. Libertinus Serif + Gill Sans is my favourite Serif and Sans pair. You can see Linux Libertine in the Wikipedia logo. Gill Sans Nova is commonly fond in the University of Tartu (Estonia) press. Although Libertinus Serif has an original Sans counterpart, its combination with Gill Sans looks most natural. I love Libertinus because of its amazingly looking ligatures, and it is also compatible with MS Equation Editor and LaTeX.
PS One can play with fonts in the EU projects to make their proposal more appealing. Like Estonian grants, I prefer calls, where applicants fill out online forms without changing the text appearance. Of course, the text looks ugly due to nasty line breaks, horrible chemical formulas and mathematical equations, and poor typography. Still, the competition is more fair because everyone is in the same conditions.
GPAW has a large set of PAW setups (updated in 2016) for elements from H to Rn, excluding lanthanides, actinides, and radioactive elements. One can generate new setups with a PAW generating build-in tool and their own risk. One can use optimized norm-conserving Vanderbilt SG15 pseudopotentials (updated in 2017) or norm-conserving Hartwigsen-Goedecker-Hutter HGH pseudopotentials (see also GPAW intro) or even JTH pseudopotentials from ABINIT. There are even more setups, including f-elements, listed on the QE webpage. The great thing about these setups is that they use a similar format – either xml or upf. Apparently, GPAW can read both formats, although there is no relevant documentation. So, there are many ways to run calculations with elements that are missing in the GPAW default setups set. QuantumATK webpage provides an overview of pseudopotentials and even suggests mixing them. I hope that in the future, these and new PAWs will be gathered together like basis sets at the basis sets exchange portal.
P.S. Interesting https://esl.cecam.org/data/ and https://molmod.ugent.be/deltacodesdft
These are undeservedly less attention to optimizers than density functionals (concerning Jacob’s ladder). It is not even covered in the recent review: Best-Practice DFT Protocols for Basic Molecular Computational Chemistry. At the same time, in my current projects, the most resource-demanding was geometry optimization – the time spent on optimizing structures was much longer than a single-point calculation. Papers that introduce new (AI-based) optimizers promise significant speed-up. However, there are always some problems:
The tested systems are different from my electrochemical interfaces.
The code is not available or difficult to install.
The code is outdated and contains bugs.
Optimizers perform worse than the common ones, like QuasiNewton in ASE.
ASE wiki lists all internal and some external optimizers and provides their comparison. I have checked the most promising on a high-entropy alloy slab.
Observation 1. QuasiNewton outperforms all other optimizers. Point. I have run a standard GPAW/DFT/PBE/PW optimization with various optimizers:
Observation 2. Pre-optimizing the slab with a cheaper method does not reduce the number of optimization steps. I have preoptimized the geometry with TBLITE/DFTB/GFN1-xTB to continue with GPAW/DFT/PBE/PW. Preoptimization takes just some minutes and the obtained geometry looks similar to the DFT one but that does not reduce the number of DFT optimization steps.
Optimizer
N steps*
Time$
N steps*#
Total time#
BFGS
16
02:44:27
17
03:01:26
LBFGS
15
02:30:35
16
02:55:04
BondMin
12
02:46:27
13
02:45:07
GPMin
12
05:26:23
31
08:14:22
MLMin
38
verylong
28
12:31:29
FIRE
38
05:06:56
44
05:56:54
QuasiNewton
8
01:36:23
9
02:00:10
Note * – the printed number of steps might different from the actuall number of calculations because each calculator has a different way of reporting that number.
Note $ – the time between the end of the first and last steps.
Note # – started from the TBLITE/DFTB/GFN1-xTB preoptimized geometry.
N.B! I have done my test only once in two runs: starting with slab.xyz and preoptized geometry. Runs were on similar nodes and all optimizations were done on the same node.
Conclusion. Do not believe in claims in articles advertizing new optimizers – Run your tests before using them.
A practical finding. The usual problem with calculations that require many optimization steps is that they need to fit into HPC time limits. On the restart, ASE usually rewrites the trajectory. Some optimizers (GPMin and AI-based) could benefit from reading the full trajectory. So, I started writing two trajectories and a restart file like this.
# Restarting
if os.path.exists(f'{name}_last.gpw') == True and os.stat(f'{name}_last.gpw').st_size > 0:
atoms,calc = restart(f'{name}_last.gpw', txt=None)
parprint(f'Restart from the gpw geometry.')
elif os.path.exists(f'{name}_full.traj') == True and os.stat(f'{name}_full.traj').st_size > 0:
atoms = read(f'{name}_full.traj',-1)
parprint(f'Restart with the traj geometry.')
else:
atoms = read(f'{name}_init.xyz')
parprint(f'Start with the initial xyz geometry.')
# Optimizing
opt = QuasiNewton(atoms, trajectory=f'{name}.traj', logfile=f'{name}.log')
traj= Trajectory(f'{name}_full.traj', 'a', atoms)
opt.attach(traj.write, interval=1)
def writegpw():
calc.write(f'{name}_last.gpw')
opt.attach(writegpw, interval=1)
opt.run(fmax=0.05, steps=42)
Here are some details on the tests.
My gpaw_opt.py for DFT calculations on 24 cores:
# Load modules
from ase import Atom, Atoms
from ase.build import add_adsorbate, fcc100, fcc110, fcc111, fcc211, molecule
from ase.calculators.mixing import SumCalculator
from ase.constraints import FixAtoms, FixedPlane, FixInternals
from ase.data.vdw_alvarez import vdw_radii
from ase.db import connect
from ase.io import write, read
from ase.optimize import BFGS, GPMin, LBFGS, FIRE, QuasiNewton
from ase.parallel import parprint
from ase.units import Bohr
from bondmin import BondMin
from catlearn.optimize.mlmin import MLMin
from dftd4.ase import DFTD4
from gpaw import GPAW, PW, FermiDirac, PoissonSolver, Mixer, restart
from gpaw.dipole_correction import DipoleCorrection
from gpaw.external import ConstantElectricField
from gpaw.utilities import h2gpts
import numpy as np
import os
atoms = read('slab.xyz')
atoms.set_constraint([FixAtoms(indices=[atom.index for atom in atoms if atom.tag in [1,2]])])
# Set calculator
kwargs = dict(poissonsolver={'dipolelayer':'xy'},
xc='RPBE',
kpts=(4,4,1),
gpts=h2gpts(0.18, atoms.get_cell(), idiv=4),
mode=PW(400),
basis='dzp',
parallel={'augment_grids':True,'sl_auto':True,'use_elpa':True},
)
calc = GPAW(**kwargs)
#atoms.calc = SumCalculator([DFTD4(method='RPBE'), calc])
#atoms.calc = calc
# Optimization paramters
maxf = 0.05
# Run optimization
###############################################################################
# 2.A. Optimize structure using MLMin (CatLearn).
initial_mlmin = atoms.copy()
initial_mlmin.set_calculator(calc)
mlmin_opt = MLMin(initial_mlmin, trajectory='results_mlmin.traj')
mlmin_opt.run(fmax=maxf, kernel='SQE', full_output=True)
# 2.B Optimize using GPMin.
initial_gpmin = atoms.copy()
initial_gpmin.set_calculator(calc)
gpmin_opt = GPMin(initial_gpmin, trajectory='results_gpmin.traj', logfile='results_gpmin.log', update_hyperparams=True)
gpmin_opt.run(fmax=maxf)
# 2.C Optimize using LBFGS.
initial_lbfgs = atoms.copy()
initial_lbfgs.set_calculator(calc)
lbfgs_opt = LBFGS(initial_lbfgs, trajectory='results_lbfgs.traj', logfile='results_lbfgs.log')
lbfgs_opt.run(fmax=maxf)
# 2.D Optimize using FIRE.
initial_fire = atoms.copy()
initial_fire.set_calculator(calc)
fire_opt = FIRE(initial_fire, trajectory='results_fire.traj', logfile='results_fire.log')
fire_opt.run(fmax=maxf)
# 2.E Optimize using QuasiNewton.
initial_qn = atoms.copy()
initial_qn.set_calculator(calc)
qn_opt = QuasiNewton(initial_qn, trajectory='results_qn.traj', logfile='results_qn.log')
qn_opt.run(fmax=maxf)
# 2.F Optimize using BFGS.
initial_bfgs = atoms.copy()
initial_bfgs.set_calculator(calc)
bfgs_opt = LBFGS(initial_bfgs, trajectory='results_bfgs.traj', logfile='results_bfgs.log')
bfgs_opt.run(fmax=maxf)
# 2.G. Optimize structure using BondMin.
initial_bondmin = atoms.copy()
initial_bondmin.set_calculator(calc)
bondmin_opt = BondMin(initial_bondmin, trajectory='results_bondmin.traj',logfile='results_bondmin.log')
bondmin_opt.run(fmax=maxf)
# Summary of the results
###############################################################################
fire_results = read('results_fire.traj', ':')
parprint('Number of function evaluations using FIRE:',
len(fire_results))
lbfgs_results = read('results_lbfgs.traj', ':')
parprint('Number of function evaluations using LBFGS:',
len(lbfgs_results))
gpmin_results = read('results_gpmin.traj', ':')
parprint('Number of function evaluations using GPMin:',
gpmin_opt.function_calls)
bfgs_results = read('results_bfgs.traj', ':')
parprint('Number of function evaluations using BFGS:',
len(bfgs_results))
qn_results = read('results_qn.traj', ':')
parprint('Number of function evaluations using QN:',
len(qn_results))
catlearn_results = read('results_mlmin.traj', ':')
parprint('Number of function evaluations using MLMin:',
len(catlearn_results))
bondmin_results = read('results_bondmin.traj', ':')
parprint('Number of function evaluations using BondMin:',
len(bondmin_results))
My tblite_opt.py for DFTB calcualation with just one core. It takes some minutes but eventually crashes 🙁
# Load modules
from ase import Atom, Atoms
from ase.build import add_adsorbate, fcc100, fcc110, fcc111, fcc211, molecule
from ase.calculators.mixing import SumCalculator
from ase.constraints import FixAtoms, FixedPlane, FixInternals
from ase.data.vdw_alvarez import vdw_radii
from ase.db import connect
from ase.io import write, read
from ase.optimize import BFGS, GPMin, LBFGS, FIRE, QuasiNewton
from ase.parallel import parprint
from ase.units import Bohr
from tblite.ase import TBLite
import numpy as np
import os
# https://tblite.readthedocs.io/en/latest/users/ase.html
atoms = read('slab.xyz')
atoms.set_constraint([FixAtoms(indices=[atom.index for atom in atoms if atom.tag in [1,2]])])
# Set calculator
calc = TBLite(method="GFN1-xTB",accuracy=1000,electronic_temperature=300,max_iterations=300)
atoms.set_calculator(calc)
qn_opt = QuasiNewton(atoms, trajectory='results_qn.traj', logfile='results_qn.log', maxstep=0.1)
qn_opt.run(fmax=0.1)
To compare structures I have used MDanalysis, which unfortunately does not work with ASE traj, so I prepared xyz-files with “ase convert -n -1 file.traj file.xyz”
Between installation with conda and compilation of libraries, an intermediate path – installation of GPAW with pip – is a compromise for those who wish to text specific GPAW branches or packages.
For example, I wish to text self-interaction error correction (SIC) and evaluate Bader charges with pybader. Neither SIC nor pybader is compatible with the recent GPAW. Here is not to get a workable version.
# numba in pybader is not compatible with python 3.11, so create a conda environment with python 3.10
conda create -n gpaw-pip python=3.10
conda activate gpaw-pip
conda install -c conda-forge libxc libvdwxc
conda install -c conda-forge ase
# ensure that you install the right openmpi (not external)
conda install -c conda-forge openmpi ucx
conda install -c conda-forge compilers
conda install -c conda-forge openblas scalapack
conda install -c conda-forge pytest
pip install pybader
# Get a developer version of GPAW with SIC
git clone -b dm_sic_mom_update https://gitlab.com/alxvov/gpaw.git
cd gpaw
cp siteconfig_example.py siteconfig.py
# In the siteconfig.py rewrite
'''
fftw = True
scalapack = True
if scalapack:
libraries += ['scalapack']
'''
unset CC
python -m pip install -e .
gpaw info
While preparing the final report on the past MSCA project, I found some memorable pictures. Here me, my wife and nephew are building a LEGO illustration for the project proposal. Yes, we had some fun while I was thinking about the concept.
The results looks pretty.
Still, as the concept illustration, I draw this figure. Today, I have reused it for the report illustration.
For a long time I wanted to see ASE atoms in my Jupyter notebook. My previous attempts were usually unsuccessful. Today I decided to try again. First ASE wiki suggests x3d and webngl:
By the way, the model is from my “Surface Curvature Effect on Dual-Atom Site Oxygen Electrocatalysis” paper, which you can read at chemRxiv until it turns Gold Open Access.
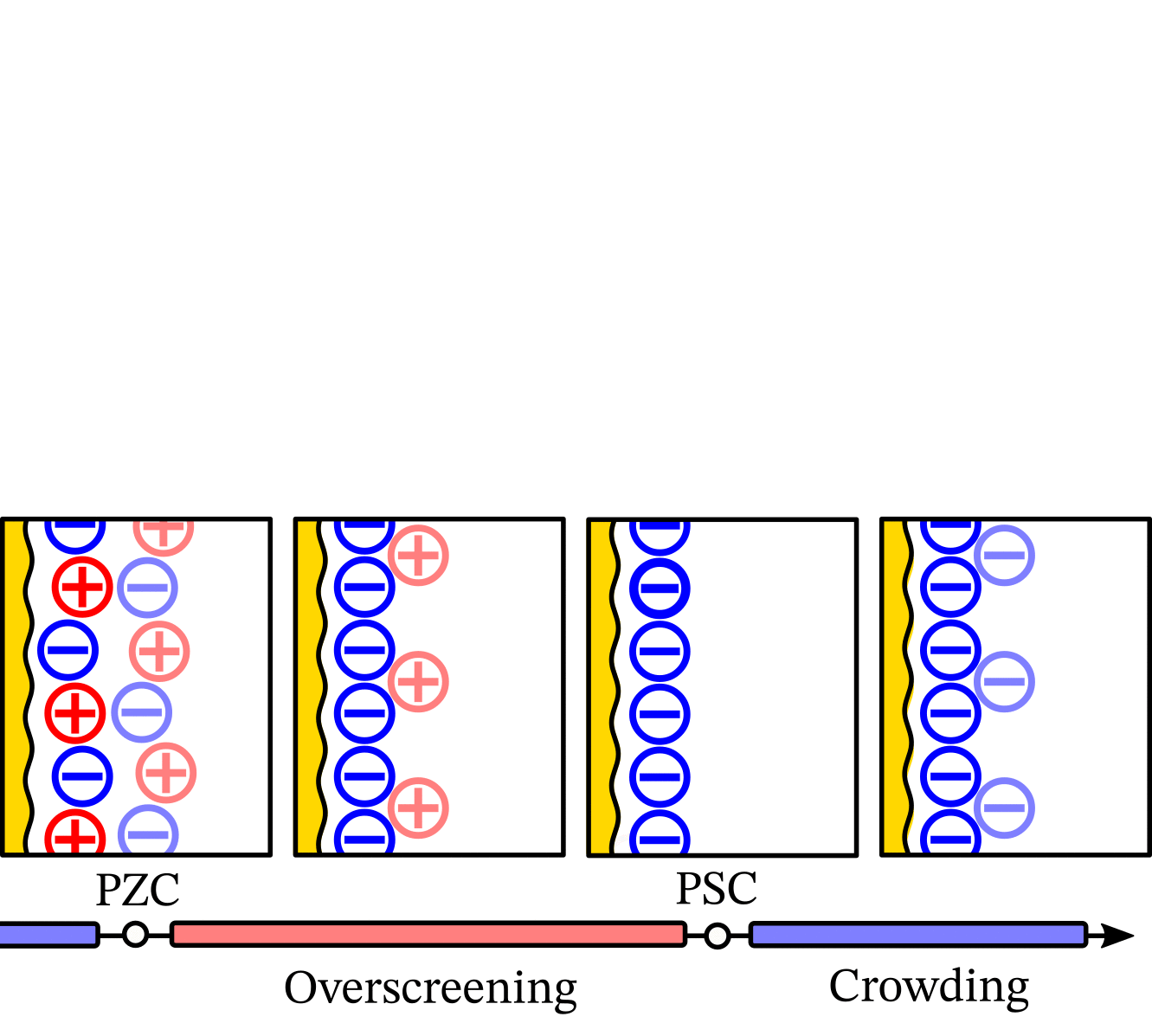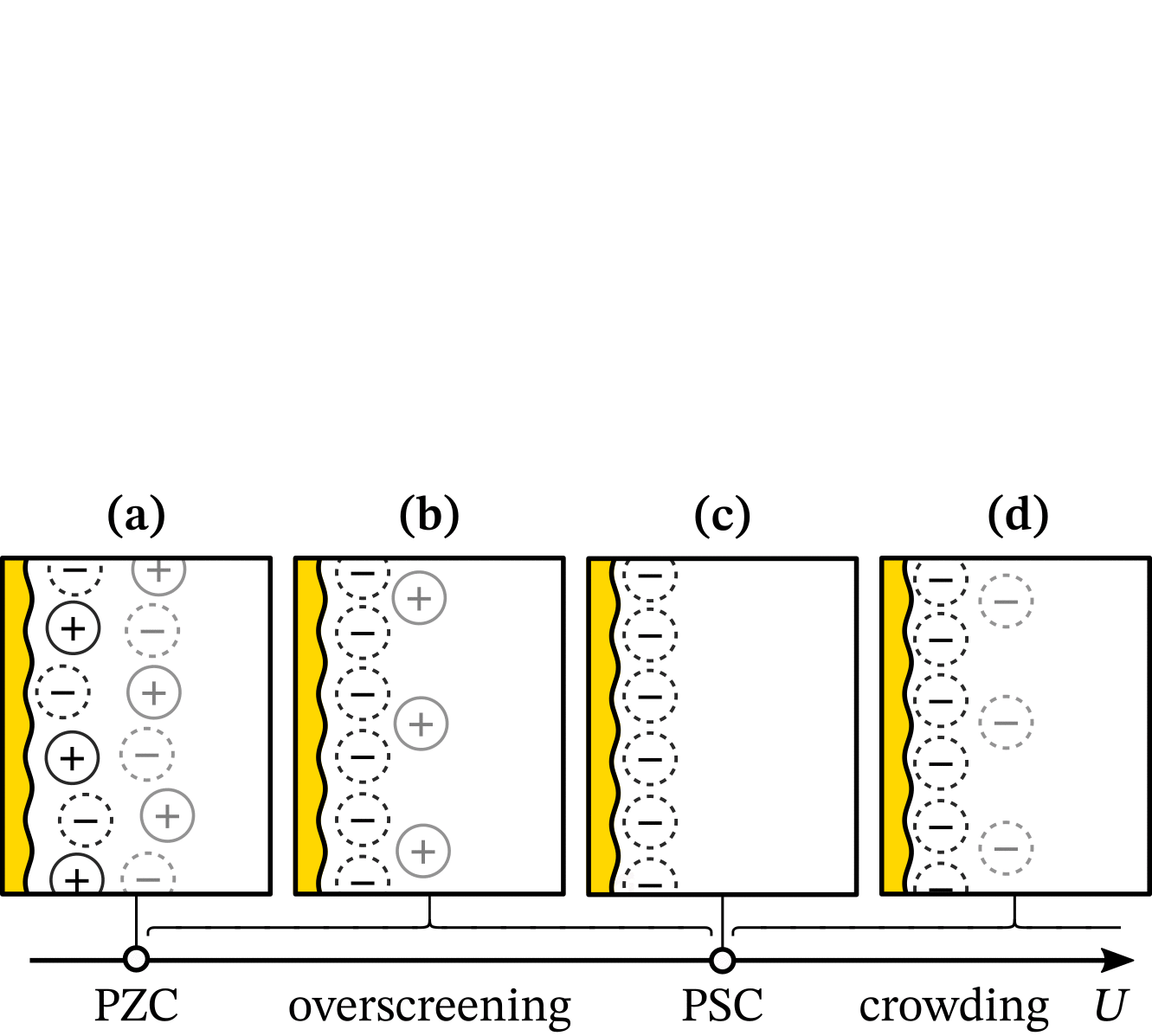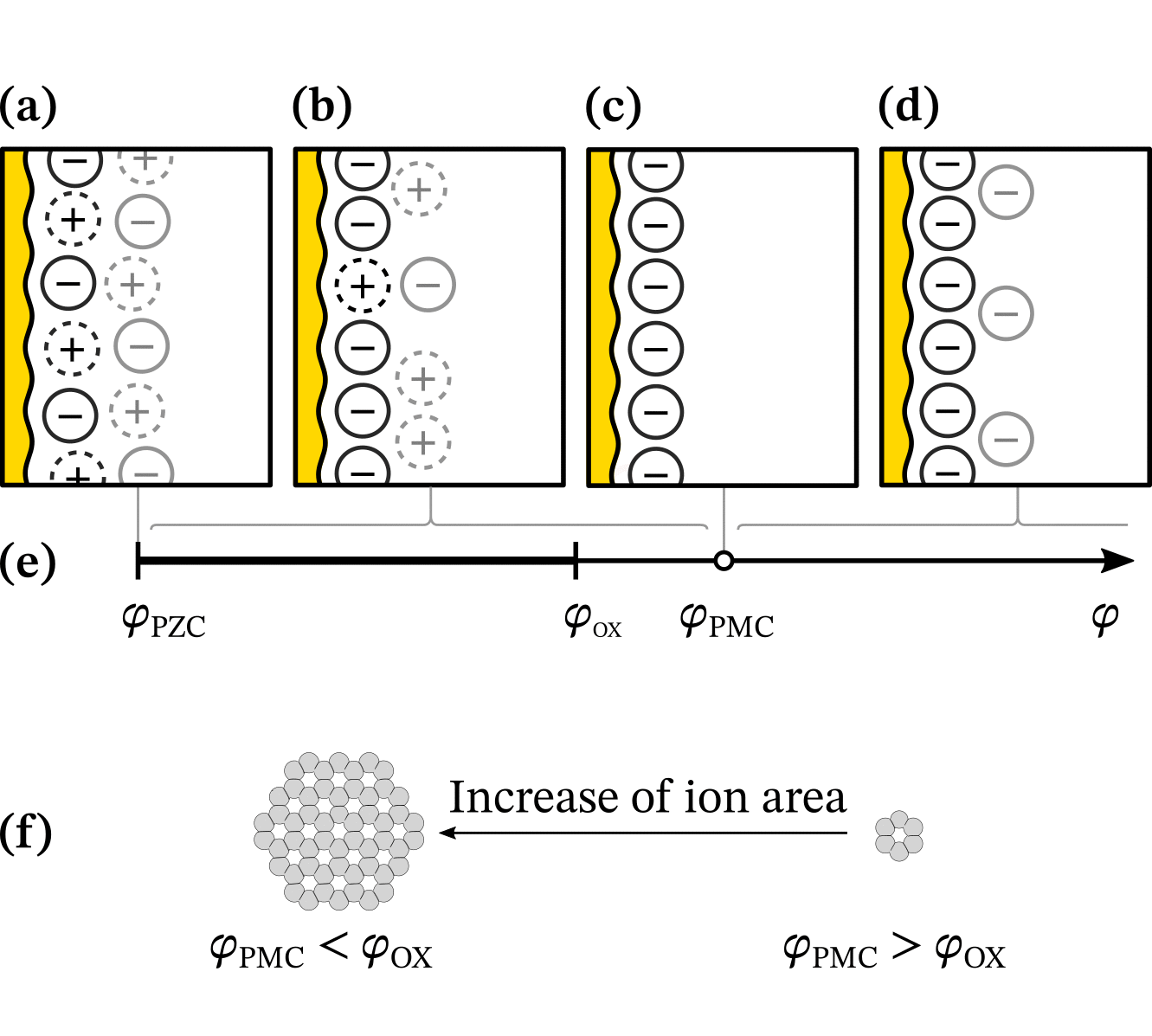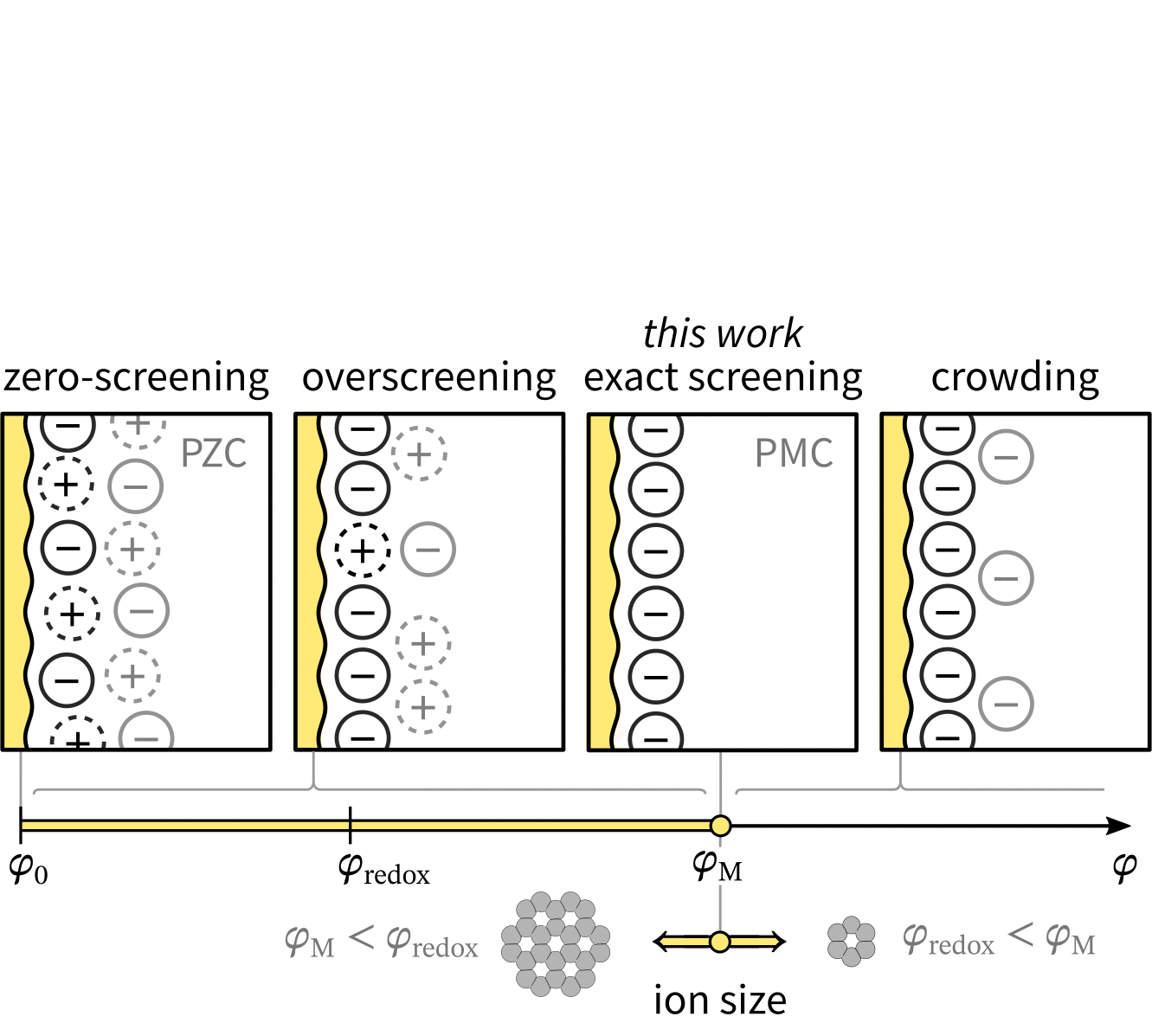
With chatGPT, we created a wavy electrode and arranged ions in Matplotlib. Then, we created over 30 versions in Inkscape, as shown in this animation (also made with guidance from chatGPT):
It was crucial to have versions instead of working on the same file because we often reverted to previous versions after trying new visual elements. Our final figure illustrates that the Potential of Monolayer Charge (PMC) is right between overscreening and crowding regimes. Moreover, the PMC can fit into the electrochemical stability window by lowering its absolute value with varying ion area. Read more in the “Potential of monolayer charge” letter.
Here are two examples of lyrics generated by ChatGPT. To make it even more fun, copy-paste the text to deepl.com (better than google translate) and make it read/sign the lyrics. melobytes is not good at all.
Verse 1: Carbon in the air, causing so much harm But we can make a change, with carbon capture Trapping it away, before it can do more We can reduce the harm, and protect our shore
Chorus: Carbon capture, carbon capture A solution for our planet’s future Carbon capture, carbon capture Together we can make it happen
Verse 2: Technology is key, to making it a success We can power our lives, while reducing the excess Carbon in the air, will no longer be a threat With carbon capture, we can make a new bet
Chorus: Carbon capture, carbon capture A solution for our planet’s future Carbon capture, carbon capture Together we can make it happen
Bridge: We can make a difference, with every step we take Towards a brighter future, for the sake of those who’ll come Let’s work together, and make this dream come true With carbon capture, we can make a better future for you
Chorus: Carbon capture, carbon capture A solution for our planet’s future Carbon capture, carbon capture Together we can make it happen.
and another one
Electricity, oh electricity Flowing through a circuit so free But when it comes to ionic liquids There’s more to it, as you’ll see
The electric double layer Is a fascinating thing It forms between the electrodes And the ionic liquids they bring
The ions in the liquid Are attracted to the metal They line up in a layer It’s really quite essential
This double layer of charge Controls the flow of electricity It’s a key part of the circuit That makes our technology so advanced, you see
So next time you flip a switch Or plug in your phone to charge Think of the electric double layer Making it all possible, oh so large!
Archives
Categories
My work was supported by the Estonian Research Council under grants PUT1107, PRG259 and STP52. My research was supported by the from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 101031656. All related posts are tagged with MSCA.