For me codex is a form of LLM that can read and edit my files. Today, with my friend Maksim Mishin we tested it on several of my current work tasks. One particular example resonates with my very first try of an LLM from December 2022: https://doublelayer.eu/vilab/2022/12/01/playing-with-galactica/
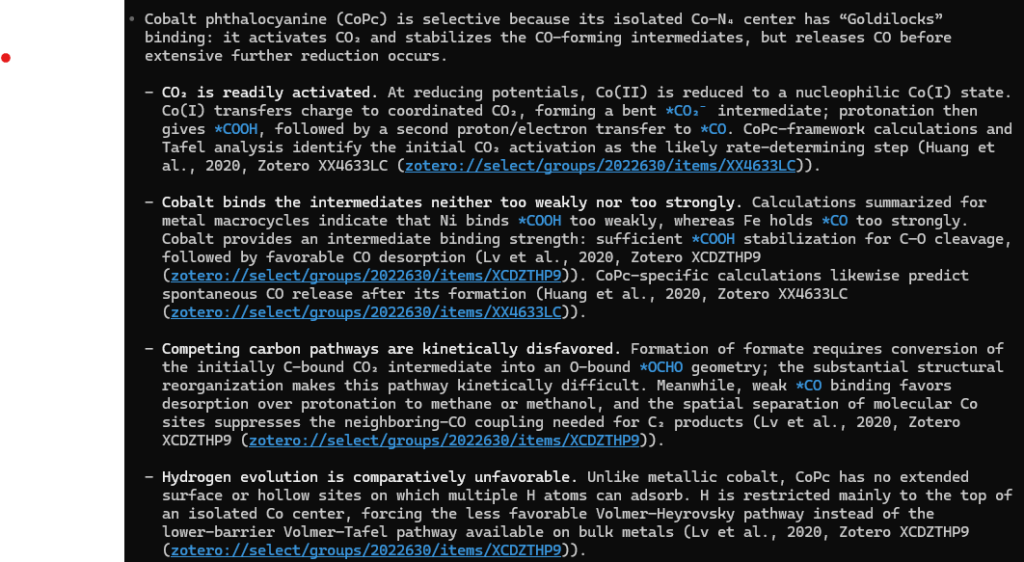
Here is where we are after 3.5 years. An agent with skills enabling interaction with my Zotero library allows to answer a questions like why CoPc is so selective towards CO in CO2RR.
So I have asked ScopusAI, ChatGPT deepsearch, and Google Scholar Labs the same question. Google Scholar Labs are the fastest and rather good, i.e. gives meaningful and diverse explanations. ScopusAI picked a very non-standard explanation and presented it in different ways in its long answer – a highly doubtful result. ChatGPT deepsearch takes too long!!! In 8 minutes it generated a report which is surprisingly deep and correct.
I am repeating the text with ScopusAI, ChatGPT deepsearch, and Google Scholar Labs at least twice a year. ScopusAI consistently gives results that differ from my expert (biassed) knowledge, so I would not trust it and, thus, do not recommend it to my colleagues. Google Scholar Labs is a cool tool to pre-select sources. ChatGPT deepsearch is at least progressing – it is the first time it gives a meaningful overview.
Overall, the experiment with Codex feels different. Here it helps me to summarize already pre-selected sources. And it allows me to check them in one click. Which makes this approach at least promising.
For a project idea I want to draw a kaleidoscope. Apparently LLM cannot do that. So with chatGPT we wrote this code to play with. This produces something like this:
import math
import random
from PIL import Image, ImageDraw
W = H = 1400
cx = cy = W / 2
R = 520
gap = 12
random.seed(4)
img = Image.new("RGBA", (W, H), "white")
draw = ImageDraw.Draw(img)
palette = [
(115, 45, 185),
(35, 85, 215),
(0, 180, 220),
(0, 165, 90),
(150, 205, 0),
(255, 210, 0),
(255, 130, 0),
]
def color_by_radius(r):
i = min(int(r / R * len(palette)), len(palette) - 1)
return palette[i]
def inside_circle(poly, radius=R):
return all(math.hypot(x, y) <= radius for x, y in poly)
def centroid(poly):
return (
sum(p[0] for p in poly) / len(poly),
sum(p[1] for p in poly) / len(poly)
)
def rotate_poly_about_centroid(poly, deg):
mx, my = centroid(poly)
a = math.radians(deg)
return [
(
mx + (x - mx) * math.cos(a) - (y - my) * math.sin(a),
my + (x - mx) * math.sin(a) + (y - my) * math.cos(a)
)
for x, y in poly
]
def scale_poly_about_centroid(poly, factor):
mx, my = centroid(poly)
return [
(
mx + (x - mx) * factor,
my + (y - my) * factor
)
for x, y in poly
]
def rot_point(p, deg):
x, y = p
a = math.radians(deg)
return (
x * math.cos(a) - y * math.sin(a),
x * math.sin(a) + y * math.cos(a)
)
def to_screen(p):
x, y = p
return (cx + x, cy - y)
def mirror_y(poly):
return [(-x, y) for x, y in poly]
def shrink(poly, amount):
mx, my = centroid(poly)
out = []
for x, y in poly:
vx, vy = x - mx, y - my
d = math.hypot(vx, vy)
if d == 0:
out.append((x, y))
else:
out.append((x - amount * vx / d, y - amount * vy / d))
return out
def draw_rounded_triangle(draw, pts, radius, fill, steps=10):
rounded = []
for i in range(3):
p_prev = pts[(i - 1) % 3]
p = pts[i]
p_next = pts[(i + 1) % 3]
d1 = math.dist(p, p_prev)
d2 = math.dist(p, p_next)
if d1 == 0 or d2 == 0:
continue
t1 = min(radius / d1, 0.45)
t2 = min(radius / d2, 0.45)
a = (
p[0] + (p_prev[0] - p[0]) * t1,
p[1] + (p_prev[1] - p[1]) * t1
)
b = (
p[0] + (p_next[0] - p[0]) * t2,
p[1] + (p_next[1] - p[1]) * t2
)
for j in range(steps + 1):
u = j / steps
qx = (1 - u) ** 2 * a[0] + 2 * (1 - u) * u * p[0] + u ** 2 * b[0]
qy = (1 - u) ** 2 * a[1] + 2 * (1 - u) * u * p[1] + u ** 2 * b[1]
rounded.append((qx, qy))
if len(rounded) >= 3:
draw.polygon(rounded, fill=fill)
def draw_triangle(poly, corner_radius):
if not inside_circle(poly):
return
r = sum(math.hypot(x, y) for x, y in poly) / 3
p = shrink(poly, gap)
pts = [to_screen(q) for q in p]
draw_rounded_triangle(
draw,
pts,
radius=corner_radius,
fill=color_by_radius(r),
steps=12
)
s = 54
h = s * math.sqrt(3) / 2
base = []
for row in range(14):
y = 45 + row * h
max_x = math.tan(math.radians(30)) * y
cols = int(max_x / s) + 3
for col in range(cols):
x = col * s + (row % 2) * s / 2
tri_up = [
(x, y),
(x + s, y),
(x + s / 2, y + h)
]
tri_dn = [
(x + s, y),
(x + 1.5 * s, y + h),
(x + s / 2, y + h)
]
for tri in (tri_up, tri_dn):
c_x, c_y = centroid(tri)
theta = math.degrees(math.atan2(c_x, c_y))
r = math.hypot(c_x, c_y)
if 0 <= theta <= 30 and 25 < r < R:
tri = scale_poly_about_centroid(tri, random.uniform(0.80, 2.00)) # VARIABLE
tri = rotate_poly_about_centroid(tri, random.uniform(-30, 30)) #VARIABLE
if inside_circle(tri):
corner_radius = random.uniform(10, 100) #VARIABLE
base.append((tri, corner_radius))
for k in range(6):
angle = k * 60
for tri, corner_radius in base:
tri1 = [rot_point(p, angle) for p in tri]
tri2 = [rot_point(p, angle) for p in mirror_y(tri)]
draw_triangle(tri1, corner_radius)
draw_triangle(tri2, corner_radius)
img.save("kaleidoscope_variable_rounding.png")
Then we thought a bit more. My ideas was to stop drawing and use symbols instaed. We came up with another code producing something like this:
Or this (with the right fonts):
import math
import random
from PIL import Image, ImageDraw, ImageFont
def draw_symbol(symbol, x, y, size, angle, color):
font = ImageFont.truetype(
#"/usr/share/fonts/truetype/noto/NotoColorEmoji.ttf",
"/usr/share/fonts/truetype/noto/NotoSansSymbols2-Regular.ttf",
int(size)
)
# =========================
# Variables
# =========================
W = 1400
H = 1400
CX = W / 2
CY = H / 2
R = 560
SEED = 8
OUTPUT_FILE = "kaleidoscope_symbols_symmetric.png"
N_OBJECTS = 53
WEDGE_DEG = 30 # one mirror wedge
SECTOR_DEG = 60 # one full sector after mirroring
N_SECTORS = 6 # six-fold symmetry
R_MIN = 30
R_MAX = R - 35
SIZE_MIN = 30
SIZE_MAX = 120
SIZE_RANDOM = 0.25 # ±25%
ROT_RANDOM = 35 # ±35 degrees
SYMBOLS = [
"★", "♥", "◆", "●"
]
PALETTE = [
(115, 45, 185), # violet
(35, 85, 215), # blue
(0, 180, 220), # cyan
(0, 165, 90), # green
(150, 205, 0), # yellow-green
(255, 210, 0), # yellow
(255, 130, 0), # orange
]
# =========================
# Setup
# =========================
random.seed(SEED)
img = Image.new("RGBA", (W, H), "white")
draw = ImageDraw.Draw(img)
font = ImageFont.load_default()
# =========================
# Helper functions
# =========================
def color_by_radius(r):
i = min(int(r / R * len(PALETTE)), len(PALETTE) - 1)
return PALETTE[i]
def size_by_radius(r):
t = r / R
size = SIZE_MIN + (SIZE_MAX - SIZE_MIN) * t
size *= random.uniform(1 - SIZE_RANDOM, 1 + SIZE_RANDOM)
return size
def polar_local(r, theta_deg):
# local coordinate system:
# theta = 0 is the central symmetry axis of one 60-degree sector
a = math.radians(theta_deg)
return (
r * math.sin(a),
r * math.cos(a)
)
def rotate_xy(x, y, angle_deg):
a = math.radians(angle_deg)
return (
x * math.cos(a) - y * math.sin(a),
x * math.sin(a) + y * math.cos(a)
)
def to_screen(x, y):
return (
CX + x,
CY - y
)
def inside_circle_xy(x, y, margin=0):
return math.hypot(x, y) <= R - margin
def draw_symbol(symbol, x, y, size, angle, color):
font = ImageFont.truetype(font_path, int(size))
canvas = int(size * 3)
tmp = Image.new("RGBA", (canvas, canvas), (255, 255, 255, 0))
td = ImageDraw.Draw(tmp)
bbox = td.textbbox((0, 0), symbol, font=font)
tw = bbox[2] - bbox[0]
th = bbox[3] - bbox[1]
scale = max(1, int(size / 8))
td.text(
(
canvas / 2 - tw / 2,
canvas / 2 - th / 2
),
symbol,
font=font,
fill=color + (255,)
)
tmp = tmp.rotate(angle, expand=True, resample=Image.Resampling.BICUBIC)
sx, sy = to_screen(x, y)
img.alpha_composite(
tmp,
(
int(sx - tmp.width / 2),
int(sy - tmp.height / 2)
)
)
def transform_from_wedge(r, theta, sector_index, mirror):
# seed wedge: theta in [0, 30]
# mirror wedge: theta in [-30, 0]
# together: one 60-degree sector centered on 0
local_theta = theta if not mirror else -theta
x, y = polar_local(r, local_theta)
# rotate complete mirrored sector around center
x, y = rotate_xy(x, y, sector_index * SECTOR_DEG)
return x, y, local_theta + sector_index * SECTOR_DEG
# =========================
# Build seed wedge only once
# =========================
seed_objects = []
for _ in range(N_OBJECTS):
r = random.uniform(R_MIN, R_MAX)
theta = random.uniform(0, WEDGE_DEG)
symbol = random.choice(SYMBOLS)
size = size_by_radius(r)
rotation = random.uniform(-ROT_RANDOM, ROT_RANDOM)
color = color_by_radius(r)
seed_objects.append({
"r": r,
"theta": theta,
"symbol": symbol,
"size": size,
"rotation": rotation,
"color": color,
})
# =========================
# Draw reflected and rotated copies
# =========================
for sector_index in range(N_SECTORS):
for obj in seed_objects:
for mirror in [False, True]:
x, y, symmetry_angle = transform_from_wedge(
r=obj["r"],
theta=obj["theta"],
sector_index=sector_index,
mirror=mirror
)
if not inside_circle_xy(x, y, margin=obj["size"] * 0.5):
continue
rotation = obj["rotation"]
if mirror:
rotation = -rotation
rotation += symmetry_angle
draw_symbol(
symbol=obj["symbol"],
x=x,
y=y,
size=obj["size"],
angle=rotation,
color=obj["color"]
)
# =========================
# Circular crop
# =========================
mask = Image.new("L", (W, H), 0)
mask_draw = ImageDraw.Draw(mask)
mask_draw.ellipse(
(CX - R, CY - R, CX + R, CY + R),
fill=255
)
out = Image.new("RGBA", (W, H), "white")
out.paste(img, (0, 0), mask)
out.save(OUTPUT_FILE)
Below is a photo of a laboratory at KongiLab – my partner-experimentalists. This is the lab where most of the experiments on CO₂ capture have been conducted over the last 3 years. After 2 years of multiple revisions, our work has just been accepted for publication. With this post, I am greeting Iuliia Vetik and congratulating all collaborators.
MSCA is often seen as a decision point.
Before the result, many think:
this will define my future.
After the result, some think:
now I am safe.
But both views come from a fixed mindset,
which leads to insecurity feeling closer to the fellowship end.
Statistically, MSCA does not define your career.
It increases your probabilities to be employed.
You are not selecting just one path.
You are expanding the number of possible paths.
These ideas come from the growth mindset framework –
it explains how mindset prepares people for success.
The people on this slide followed it consistently,
often without naming it.
So the question is not whether you win or lose the MSCA PF.
The question is how you use this step to move forward.
Let us take a look at how careers in biology
in the US careers evolve in numbers.
These are shown in black numbers.
If we add MSCA data in colour numbers,
we see a similar pattern.
Right after the fellowship,
more than half are employed.
After two years, almost 90% are employed.
And almost all say the skills gained are useful.
Herewith, the most used skills are: communication, networking, and project management.
Around 70% stay in academia, about 20% go to industry.
But only one in three in academia have permanent positions, while in industry, four out of five are permanent.
At the same time, about half of researchers desire to become a professor, but only one in ten reach that path.
So there is a clear gap between expectations and probabilities.
This picture shows that careers are not a single predefined path.
They are a set of probable stepwise paths.
Thus, the rational approach is to align expectations with probabilities to step up,
not with initial desires.
Please think on this idea.
Fixing on “precious” results is destructive.
It creates pressure before MSCA,
and false security after it.
Growing along your career,
with purpose, is constructive.
It means using each step,
not depending on a single result.
MSCA is one of many steps.
Not the destination.
So keep calm, and keep going.
Join the DoubleLayer hub as a postdoctoral fellow at the University of Latvia!
If you are a post-doc seeking independence through training skills & gaining knowledge in a supportive environment, then this post is for you. That is an opportunity to advance your career through Marie Skłodowska-Curie Actions (MSCA) by focusing on competencies – academic writing, research methods, and supervision – essential for succeeding in academia and industry.
You need to submit just one proposal on 9 September 2026 to participate in at least three funding calls. The proposal is only 10 pages long. The first application is for the MSCA or MSCA4Ukraine postdoctoral fellowship, which provides funding for up to 24 months of research and training. To get this prestigious grant, one must gain more than 96% in the evaluation. However, passing the 85% threshold already opens the opportunity to be funded through the ERA Fellowship. Moreover, passing the 80% threshold makes you eligible to be funded by the state of Latvia. Submitting to all these 3! opportunities per 1 proposal increases your chances of fulfilling your research idea and advancing your career. These fellowships include funds for salary, mobility, research, and allowances described in the following table.
Disclaimer: The data in the table might be incorrect. CCC value is taken form 2026–2027 program. Salary is estimated using this calculator.
Research experience related to the electrical double layer, see list of publications.
Experience in supervising dozens of postdocs and BSc–PhD students as well as mentoring over 100 members of the Estonian national team at the international Chemistry Olympiad.
In the 2024 MSCA call, I consulted for 5 applicants: 3 received the MSCA PF, and 2 received the Seal of Excellence. In 2025, I consulted 8 applicants: 3 received ERA PF (one eventually got MSCA), and 2 received the seal of excellence.
Further details
To be considered for the opportunity, you will undergo a pre-selection process based on your CV, project idea, and motivation letter. There are three main eligibility requirements:
You must hold a PhD and up to 8 years of full-time research experience by the time of the application. Check the eligibility calculator.
Applicants of any nationality are welcome, but they must not have lived or worked in Latvia for more than 12 months during the 3 years leading up to the closing date of the call on 10 September 2025.
Applicants must choose the Chemistry Department at the University of Latvia as their host institution.
For any other further questions, please contact vladislav.ivanistsev@doublelayer.eu. Prefix your email subject title with “DoubleLayer hub:”
The best way to present a 3D model in a figure is an isometric projection. I use it whenever I draw atomic structures or simulation cells because it is easy to comprehend.
In perspective rendering, objects farther from the camera appear smaller. That is useful in photography, but it is often misleading in scientific figures. Equal lengths should look equal. Parallel edges should remain parallel. In an isometric projection this is exactly what happens: the scale is the same along all three spatial directions.
The idea comes from technical drawing and engineering graphics in the 19th century. Engineers needed a way to draw three-dimensional machines on paper while preserving measurable proportions. The solution was to project the object along the direction of a cube’s body diagonal. When viewed this way, the three Cartesian axes appear symmetrically separated by 120°. As a result, edges parallel to those axes appear with equal foreshortening.
This produces the familiar look of isometric drawings: vertical lines remain vertical, and the other two axes appear as lines tilted by 30° from the horizontal.
For scientific graphics this view is ideal. It preserves symmetry, keeps dimensions comparable, and produces consistent figures across different structures.
I use the following rotations to produce an isometric view in ASE.
Rotate the structure:
x = 225° y = 215.264° z = 30°
The first two rotations orient the model so the viewing direction is along the cube body diagonal. The last rotation only rotates the image in the plane so that the projected axes appear at ±30°.
Example:
from ase import Atoms
from ase.io import write
import numpy as np
atoms = Atoms('H', positions=[[0, 0, 0]], cell=[20, 5, 10], pbc=False)
atoms.center()
atoms.rotate(225, 'x', rotate_cell=True)
atoms.rotate(180 + np.degrees(np.arctan(1 / np.sqrt(2))), 'y', rotate_cell=True)
atoms.rotate(30, 'z', rotate_cell=True)
write('iso.png', atoms, show_unit_cell=2)
The university of Latvia supports publishing in the following journals (as for 2025 and filtered). You must be the first or corresponding author. Write to zd@lu.lv.
Chemistry • ACS Nano • Advanced functional materials • Advanced materials • Applied catalysis B: environmental • Chemical reviews • Chemical science • Chemical society reviews • Chemistry of materials • Corrosion science • Green chemistry • Journal of catalysis • Journal of physical chemistry letters • Nano letters • Nanoscale • Nature chemistry • ACS catalysis • Advanced energy materials • Nano energy • ACS central science • Chem • 2D materials Physics • Advances in physics • Applied physics letters • Atmospheric chemistry and physics • Physical review letters • Physical review X • Physics letters B • Physics reports • Nature energy • Nature electronics • npj quantum materials
Cross-area (chemistry + physics + materials) • Energy • Energy and environmental science • Nature communications • Science • Science advances • Proceedings of the National Academy of Sciences (cross-disciplinary) • Joule • Nature sustainability
Archives
Categories
My work was supported by the Estonian Research Council under grants PUT1107, PRG259 and STP52. My research was supported by the from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 101031656. All related posts are tagged with MSCA.