For me codex is a form of LLM that can read and edit my files. Today, with my friend Maksim Mishin we tested it on several of my current work tasks. One particular example resonates with my very first try of an LLM from December 2022: https://doublelayer.eu/vilab/2022/12/01/playing-with-galactica/
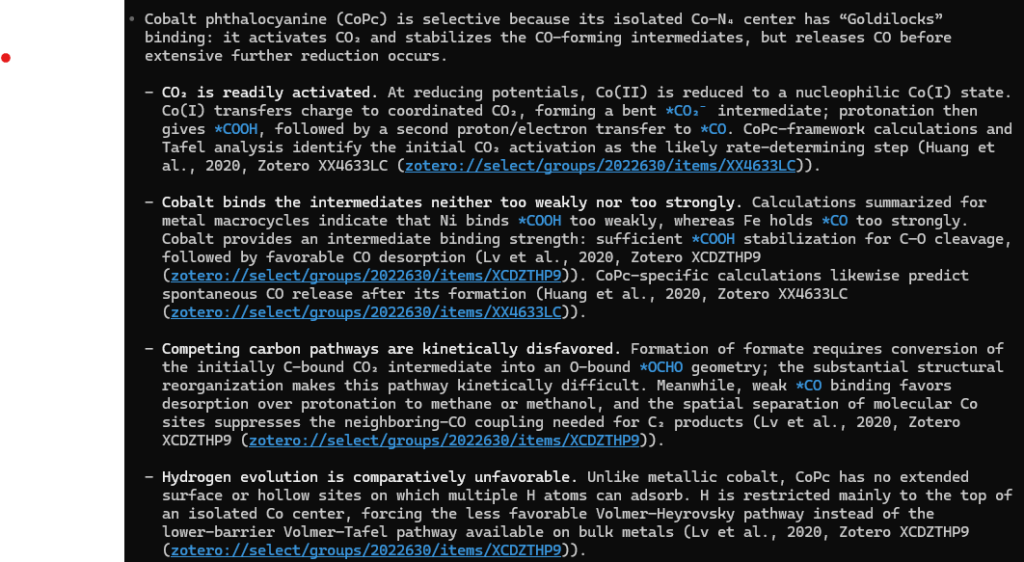
Here is where we are after 3.5 years. An agent with skills enabling interaction with my Zotero library allows to answer a questions like why CoPc is so selective towards CO in CO2RR.
So I have asked ScopusAI, ChatGPT deepsearch, and Google Scholar Labs the same question. Google Scholar Labs are the fastest and rather good, i.e. gives meaningful and diverse explanations. ScopusAI picked a very non-standard explanation and presented it in different ways in its long answer – a highly doubtful result. ChatGPT deepsearch takes too long!!! In 8 minutes it generated a report which is surprisingly deep and correct.
I am repeating the text with ScopusAI, ChatGPT deepsearch, and Google Scholar Labs at least twice a year. ScopusAI consistently gives results that differ from my expert (biassed) knowledge, so I would not trust it and, thus, do not recommend it to my colleagues. Google Scholar Labs is a cool tool to pre-select sources. ChatGPT deepsearch is at least progressing – it is the first time it gives a meaningful overview.
Overall, the experiment with Codex feels different. Here it helps me to summarize already pre-selected sources. And it allows me to check them in one click. Which makes this approach at least promising.
The best way to present a 3D model in a figure is an isometric projection. I use it whenever I draw atomic structures or simulation cells because it is easy to comprehend.
In perspective rendering, objects farther from the camera appear smaller. That is useful in photography, but it is often misleading in scientific figures. Equal lengths should look equal. Parallel edges should remain parallel. In an isometric projection this is exactly what happens: the scale is the same along all three spatial directions.
The idea comes from technical drawing and engineering graphics in the 19th century. Engineers needed a way to draw three-dimensional machines on paper while preserving measurable proportions. The solution was to project the object along the direction of a cube’s body diagonal. When viewed this way, the three Cartesian axes appear symmetrically separated by 120°. As a result, edges parallel to those axes appear with equal foreshortening.
This produces the familiar look of isometric drawings: vertical lines remain vertical, and the other two axes appear as lines tilted by 30° from the horizontal.
For scientific graphics this view is ideal. It preserves symmetry, keeps dimensions comparable, and produces consistent figures across different structures.
I use the following rotations to produce an isometric view in ASE.
Rotate the structure:
x = 225° y = 215.264° z = 30°
The first two rotations orient the model so the viewing direction is along the cube body diagonal. The last rotation only rotates the image in the plane so that the projected axes appear at ±30°.
Example:
from ase import Atoms
from ase.io import write
import numpy as np
atoms = Atoms('H', positions=[[0, 0, 0]], cell=[20, 5, 10], pbc=False)
atoms.center()
atoms.rotate(225, 'x', rotate_cell=True)
atoms.rotate(180 + np.degrees(np.arctan(1 / np.sqrt(2))), 'y', rotate_cell=True)
atoms.rotate(30, 'z', rotate_cell=True)
write('iso.png', atoms, show_unit_cell=2)
I love TeX, LaTeX and other scripting and markup languages.
I have strong feeling about all publishers! I have spend days and weeks fighting submission systems. I remember once it was about choosing between some version 2 and 3. This time I had to upload bbl file. Why can not I simply submit from overleaf.com to editorialmanager (like to arXiv)?
In overleaf it is “vital” to set your main document as the main document in the settings! – that was not intuitive at all. Still, overleaf did not show compilation files. So I had to fake submission to arXiv to download the zip with all files, including bbl, like in this instruction – that was not obvious at all.
What I have learned today is this way of submitting a LaTeX source. Start submission to arXiv, get the zip file, remove unnecessary files, upload this refined zip to editorialmanager or other platform. Save your time and nerves!
From the beginning I decided to try AI to prepare the presentation. Eventually the only to record the video turned out to be by the traditional way. Together with co-authors Ritums and Nadezda, we used PowerPoint with its slide-by-slide recording feature. As we were in 3 different locations, we exchanged the presentation several time while recording. I used chatgpt 4o and 5 to write lecturer’s notes for every slide. In particular, I gave the chat our article’s pdf-file and then discussed every slide-text using canvas-feature to polish it iteratively. Nadezda also used chatgpt to refined her slides before reading them aloud. Overall, I have spend over two weeks planning the presentation. Then a week polishing the slides. Then several days to record and re-record slides. And finally I have got this the final video-presentation.
Adding voice to a ready presentation
app.pictory.ai does a relatively good job on reading the lecturer’s notes in a ready presentation. Thought, it reads “Jan” and “OOH” in a funny way. And it adds a lot of 10–20 second pauses. Also the slide numbering is off as well as all animation. The picture is also cut from below. But overall, it takes around 2 hours to generate this voiced video and process it.
I am not responsible for the result 🙂 I have heard it and it sounds OK-ish.
Using Gemini in Google Slides
Does not work for me. Gemini wants to draw images. I just want to enter my own figures.
All I need is to convert Figures to Slides
https://www.magicslides.app promises to do exactly that but I failed with a notice that below 5 Mb files are allowed.
SlideAI extension also does not do what I want.
Ufff … manual upload is still the fastest and most robust. Well, it is not so simple, as most of my figures are in pdf, so I wrote this script to convert everything to png. When it took me 2 mins to drag-and-drop all png figure to my presentation. Hurray!
What are scaling relations in electrocatalysis, and why are they important?
Scaling relations are correlations between the adsorption energies of reaction intermediates on a catalyst’s surface. They are crucial in multi-step electrocatalytic reactions, such as the oxygen reduction reaction (ORR), carbon dioxide reduction (CO2R), and nitrogen reduction (N2RR). The concept emerged in 2005 with the discovery of linear relations between adsorption energies of intermediates like OH, OOH, and O on metal surfaces. Understanding these relations is vital because they define fundamental chemical limitations in electrocatalytic reactions, impacting the design of more efficient catalysts for energy conversion technologies like electrolysers, fuel cells, and metal-air batteries.
How do scaling relations limit the efficiency of oxygen electrocatalysis?
In oxygen electrocatalysis, particularly the oxygen reduction reaction (ORR), the adsorption energies of key intermediates (OOH, OH, O) are correlated by scaling relations. These correlations constrain the achievable catalytic activity, often visualised on “volcano plots.” The OOH-OH and O-OH scaling relations, for instance, mean that if a catalyst binds one intermediate optimally, it might bind another too strongly or too weakly, preventing it from reaching the ideal catalytic activity (the “volcano top”). This limitation is significant, as experimental results have shown catalytic overpotentials converging to a limit set by these relations for over two decades, hindering progress in sustainable energy solutions.
What are the main reaction mechanisms in oxygen electrocatalysis, and how does catalyst geometry influence them?
Oxygen electrocatalysis primarily proceeds via two mechanisms: associative and dissociative. The associative mechanism, which dominates most known catalysts, involves intermediates like OOH, OH, and O adsorbing at a single active site. Geometrically, this requires only one atom in the active site. The dissociative mechanism, conversely, requires at least two neighbouring atoms to accommodate dissociation products (O and OH). On metal surfaces, a spatial mismatch often prevents the dissociative mechanism, as O preferentially adsorbs on hollow sites and OH on top sites. However, dual-atom site catalysts (DACs) can facilitate dissociative pathways by providing two adjacent sites, allowing for the adsorption of dissociation products. The inter-atomic distance within these active sites is a critical geometric parameter that influences the energy barrier for dissociation, balancing thermodynamics and kinetics.
What is the “volcano plot” in electrocatalysis, and how do scaling relations affect it?
The “volcano plot” is a theoretical framework used to understand electrocatalysis, typically representing overpotential or activity as an “altitude” against adsorption energy descriptors. For ORR, it correlates adsorption energies with deviations from the thermodynamic equilibrium potential. Scaling relations define the “paths” or “fixed climbing routes” on this volcano plot that are accessible to catalysts. For example, the OOH-OH scaling relation appears as a plane on the three-dimensional volcano, and catalysts following this relation are confined to a specific line on the volcano’s surface. This means that while an “ideal catalyst” (the volcano’s apex) might exist theoretically, scaling relations prevent most catalysts from reaching it, limiting the search for optimal catalysts to a two-dimensional projection.
What are the five general strategies for “manipulating” scaling relations in electrocatalysis?
The review outlines five general strategies for manipulating scaling relations to enhance electrocatalytic performance:
Tuning: Adjusting the adsorption energy of a key intermediate (e.g., ∆GOH) to optimise catalyst performance within the constraints of an existing scaling relation, adhering to the Sabatier principle.
Breaking: Decreasing the intercept (β) of a scaling relation by selectively stabilising one intermediate over another (e.g., OOH relative to OH), often by introducing spectator groups that induce stabilising interactions.
Switching: Changing the slope (α) of a scaling relation by enabling an alternative reaction mechanism (e.g., switching from an associative to a dissociative mechanism in ORR) to avoid problematic intermediates. This usually requires dual active sites.
Pushing: A combined strategy that changes the slope and adjusts the intercept, simultaneously switching to an alternative mechanism and using stabilising interactions (similar to breaking).
Bypassing: Completely decoupling adsorption energies by switching between two distinct states of the catalyst (e.g., geometric or electronic) during the reaction cycle, with each state having optimal adsorption energies for specific intermediates. This strategy aims to eliminate all scaling relation constraints.
How does the “breaking” strategy specifically aim to overcome the OOH-OH scaling relation?
The “breaking” strategy focuses on reducing the intercept of the OOH-OH scaling relation (from approximately 3.2 eV to an ideal value of 2.46 eV) by selectively stabilising the OOH intermediate relative to OH. This typically involves introducing spectator groups or a second adsorption site near the active site. These spectators can form hydrogen bonds or other stabilising interactions with OOH, effectively shifting its adsorption energy without proportionally affecting OH. While challenging to achieve experimentally, this strategy has been demonstrated in oxygen evolution reactions (OER) and more recently in ORR using dual-atom catalysts (DACs) with specific active sites like PN3FeN3, where the phosphorus acts as a spectator to stabilise OOH through hydrogen bonding.
What role do Single-Atom Site Catalysts (SACs) and Dual-Atom Site Catalysts (DACs) play in manipulating scaling relations?
Single-Atom Site Catalysts (SACs) and Dual-Atom Site Catalysts (DACs) are crucial in manipulating scaling relations due to their distinct geometric and electronic properties. SACs typically allow for “on-top” adsorption, primarily favouring the associative mechanism in ORR. DACs, with their two neighbouring active sites, offer the possibility of accommodating two dissociation products simultaneously, thereby enabling the dissociative mechanism. This ability to switch mechanisms is key to the “switching” strategy, where DACs can replace the OOH intermediate with two distinct O and OH intermediates adsorbed at separate sites. Furthermore, the precise control over inter-atomic distances and curvature in DACs allows for fine-tuning of electronic structures and promoting specific interactions (like hydrogen bonding), contributing to “breaking” and “pushing” strategies.
What is the ultimate goal of manipulating scaling relations, and how does the “bypassing” strategy contribute to this vision?
The ultimate goal of manipulating scaling relations is to achieve ideal catalyst performance, ideally with zero overpotential, by overcoming the fundamental limitations imposed by these correlations. The “bypassing” strategy represents the most ambitious approach towards this goal. It seeks to completely decouple the adsorption energies of reaction intermediates by allowing the catalyst to switch between two or more distinct states (e.g., geometric, electronic, or photonic) during the reaction cycle. Each state would be optimally configured to bind specific intermediates at the ideal energy values required for efficient catalysis. While seemingly challenging in practice, this concept, inspired by natural enzymes like cytochrome c oxidase, offers a theoretical pathway to eliminate all scaling constraints and achieve the theoretical apex of the volcano plot, pushing the boundaries of what is currently achievable in electrocatalysis.
I have made so many changes to the LaTeX code that latexdiff gives 99+ errors. UPDATE: the errors were related to table and equations. To fix them I played with –math-markup=none and –add-to-config PICTURENEV=tabular, but eventually just fixed the Tables and specific errors.
In search for an alternative, have tested DiffPDF and Meld. The result looks useful, yet unsuitable for submitting them to the editor. So, this post should have a label “do not know how” instead of usual “know-how”. Still, fun.
To make the last printscreen, I did the following.
Installed packages in Linux/Ubuntu/WSL2.
sudo apt-get -y install meld calibre parallel
Saved the following code in a file named “diffdoc” (with no extensions) inside directory “/usr/local/bin”.
usage="
*** usage:
diffepub - compare text in two files. Valid format for input files are:
MOBI, LIT, PRC, EPUB, ODT, HTML, CBR, CBZ, RTF, TXT, PDF and LRS.
diffepub -h | FILE1 FILE2
-h print this message
Example:
diffepub my_file1.pdf my_file2.pdf
diffepub my_file1.epub my_file2.epub
v0.2 (added parallel and 3 files processing)
"
#parse command line options
while getopts "h" OPTIONS ; do
case ${OPTIONS} in
h|-help) echo "${usage}"; exit;;
esac
done
shift $(($OPTIND - 1))
#check if first 2 command line arguments are files
if [ -z "$1" ] || [ -z "$2" ] || [ ! -f "$1" ] || [ ! -f "$2" ]
then
echo "ERROR: input files do not exist."
echo
echo "$usage"
exit
fi
#create temporary files (first & last 10 characters of
# input files w/o extension)
file1=`basename "$1" | sed -r -e '
s/\..*$// #strip file extension
s/(^.{1,10}).*(.{10})/\1__\2/ #take first-last 10 chars
s/$/_XXX.txt/ #add tmp file extension
'`
TMPFILE1=$(mktemp --tmpdir "$file1")
file2=`basename "$2" | sed -r -e '
s/\..*$// #strip file extension
s/(^.{1,10}).*(.{10})/\1__\2/ #take first-last 10 chars
s/$/_XXX.txt/ #add tmp file extension
'`
TMPFILE2=$(mktemp --tmpdir "$file2")
if [ "$#" -gt 2 ]
then
file3=`basename "$3" | sed -r -e '
s/\..*$// #strip file extension
s/(^.{1,10}).*(.{10})/\1__\2/ #take first-last 10 chars
s/$/_XXX.txt/ #add tmp file extension
'`
TMPFILE3=$(mktemp --tmpdir "$file3")
fi
#convert to txt and compare using meld
doit(){ #to solve __space__ between filenames and parallel
ebook-convert $1
}
export -f doit
if [ "$#" -gt 2 ]
then
(parallel doit ::: "$1 $TMPFILE1" \
"$2 $TMPFILE2" \
"$3 $TMPFILE3" ) &&
(meld "$TMPFILE1" "$TMPFILE2" "$TMPFILE3")
else
(parallel doit ::: "$1 $TMPFILE1" \
"$2 $TMPFILE2" ) &&
(meld "$TMPFILE1" "$TMPFILE2")
fi
Made sure the owner is me and it has execution permissions:
Recently I noticed that my colab notebooks get too often disconnected. Yes, their run takes over an hour, which is a usual timing for a seminar. I just want to test that they run by executing all cells and leaving my workplace.
Here is how to set up an automatic reconnect using javascript functions that one can enter to the browser console:
function ConnectButton() {console.log("Working");document.querySelector("#connect").click()}
setInterval(ConnectButton,600000);
Archives
Categories
My work was supported by the Estonian Research Council under grants PUT1107, PRG259 and STP52. My research was supported by the from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 101031656. All related posts are tagged with MSCA.